As an Amazon Associate I earn from qualifying purchases.

The Food and Drug Administration (FDA) is carrying out a brand-new method to supply speculative gene treatments to clients with uncommon conditions without going through scientific trials. This structure might approve these clients access to personalized treatments, however professionals are divided over whether the regulative modification is safe enough for clients.
Dr. Senthil Bhoopalana genome-editing professional at St. Jude’s Children Research Hospital in Tennessee, stated that, although the structure is still emerging and the information need more conversation in between the general public and stakeholders, “it’s an exciting step in the right direction.”
Arthur Caplana medical ethicist at New York University, stated more pressure to allow access to brand-new treatments has actually led the FDA “to allow more risk to subjects, and more risk of failure post-approval, by being willing to accept weaker evidence.”
Before getting FDA approval, the majority of treatments need medical trials with hundreds or countless individuals to reveal a drug is safe and works. Sometimes, the company grants sped up approval for interventions that appear to reveal an advantage in little trials, when clients are extremely ill and have no other treatment choices.
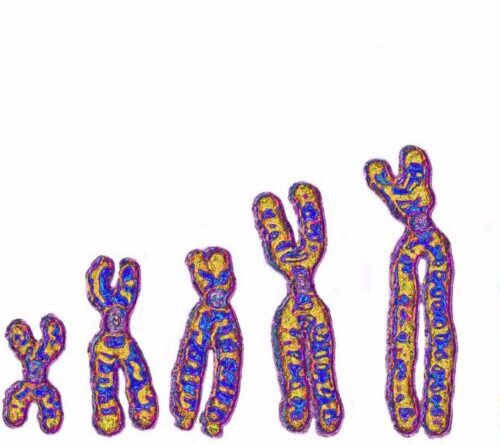
The brand-new technique, called the possible system pathwould make it possible for the FDA to give authorization to utilize treatments that have not been evaluated in human beings however might plausibly prosper.
The path would use just to particular treatments, such as gene treatments that appropriate single-letter DNA mistakes, where massive scientific trials would be difficult. Take cystic fibrosis as an example. Around 40,000 individuals in the U.S. have this condition, however numerous anomalies can trigger it, Bhoopalan stated. As an outcome, you can’t utilize one gene treatment formula to deal with every client.
Get the world’s most interesting discoveries provided directly to your inbox.
If a gene-editing tool and shipment method have actually been revealed to be safe in previous human trials, the path would enable drug designers to modify the sequence-specific component of the formula, such as a guide RNA that informs the DNA “scissors” where to fix an anomaly. The particular gene-editing tool, such as a base editor, might be personalized for particular anomalies in each cystic fibrosis client. This resembles how food manufacturers require just reveal that a component is safe as soon as before including it in numerous food products.
“It’s possible that in the fullness of time, we’ll see that they’ve lowered the bar.”
Dr. J. Paul Taylor, neurologist at St. Jude Children’s Research Hospital.
“The safety data can be extrapolated if you’re using the same delivery mechanism,” Bhoopalan stated. “You’re really only changing the guide.” If the modification you make in the body is switching a malfunctioning anomaly with the kind that healthy individuals have, you would not expect adverse effects, he included.
Caplan concurred that this specific usage of the path does not appear, on its face, to be high-risk. The security of base editors has actually been evaluated just in reasonably little trials therefore far, with no more than 15 individualsWith a sample size this little, it’s tough to reveal a provided gene treatment resulted in favorable health results. What’s more, without carrying out bigger trials including hundreds or countless individuals, it’s difficult to understand whether base editors trigger unusual adverse effects.
At least 65 small trials have actually examined using particular infections as lorries to provide liver-targeting gene treatments that deal with hemophilia. While the majority of these research studies reveal pledge, a bigger trial including 134 individuals exposed unusual adverse effects, such as raised liver enzymes, swelling and allergies.
“The level of risk doesn’t keep me awake at night, but there are unknowns,” Caplan stated. “I think it would be very important to have serious follow-ups following FDA drug approval.”
That’s where he sees the capacity for issues to sneak in. Post-approval tracking of drugs has “never been done with earnestness,” regardless of guarantees made by pharmaceutical business. “If we’re going to take more risk to go faster at the front end, you have to beef up what’s required and what’s going to be monitored at the back end, post approval.”
Still, that does not imply the level of post-approval analysis will be lower than it has actually been formerly.
“It’s possible that in the fullness of time, we’ll see that they’ve lowered the bar,” stated Dr. J. Paul Taylora neurologist who deals with hereditary neurodevelopmental conditions at St. Jude Children’s Research Hospital. “But the stated intent is not to change the level of substantial evidence [through post-approval monitoring].”
In a post released last November in The New England Journal of Medicinethe FDA laid out which requirements an illness would require to satisfy to get approved for this path. The possible system path would be dismissed for conditions with uncertain causes, such as dementia, Taylor kept in mind.
“This is great for monogenic disorders, which are caused by mutations in a single gene,” Bhoopalan stated. It would be more difficult to utilize this path for polygenic illness, which are caused by a selection of anomalies, he included, as you would need to effectively remedy several anomalies to see an advantage.
Instead of remedying a defective anomaly, gene treatment might be utilized to “switch on” a backup gene when it comes to spine muscular atrophy, Taylor stated, which is deadly in kids who do not get treatment.
“I think we have to start thinking about this as an inevitable next step.”
Dr. Senthil Bhoopalan, genome-editing specialist at St. Jude’s Children Research Hospital
There are some monogenic conditions that might not fulfill the requirement. Scattered intrinsic pontine glioma is a brain growth that appears in young kids who bring a defective gene. Taylor stated professionals are divided on whether reversing this anomaly alone might diminish the growths or if other anomalies that look like the growth establishes might continue to drive the cancer even if the preliminary anomaly were remedied.
Another FDA requirement needs physicians to verify that the client’s tissues have actually been modified. “It might be harder to quantitate when you’re editing a critical organ like the liver, because you cannot get a piece of liver and measure how much has been edited,” Bhoopalan stated.
Associated stories
Physicians might require to consistently sample tissues from clients, as research studies in mice have actually revealed that gene treatments can subside in time, recommending that some might not work as a “one-and-done” treatment. This would be much more difficult to achieve if you might just sample tissues with intrusive surgical treatment.
Some body areas may be tough to target with gene shipment systems in the very first location. The blood, bone marrow, liver and lungs might facilitate targets, Bhoopalan stated. The heart, on the other hand, might be hard to modify due to the fact that a layer of tightly-packed cells develops a barrier that stops gene treatment vectors from crossing into heart tissue.
More conversation is required to clarify which conditions can benefit from this expedited approval and how clients’ health can be kept track of later, specialists hope the brand-new path might assist individuals with unusual conditions.
“I think we have to start thinking about this as an inevitable next step,” Bhoopalan stated.
This post is for educational functions just and is not implied to use medical suggestions.
Learn more
As an Amazon Associate I earn from qualifying purchases.